How to spot zebras – pheochromocytoma
What is it?
Paragangliomas (PGL) are rare vascular tumours that originate from the paraganglia of the autonomic nervous system. They can occur anywhere from the skull base to the pelvis. The term paraganglioma is used to describe any tumour arising from the paraganglia, whether sympathetic or parasympathetic in origin. The term phaeochromocytoma (PCC) specifically describes a PGL arising in the adrenal medulla. These tumours, collectively referred to as PPGL in this article, cause morbidity and mortality through their mass effect, malignancy potential and secretory effects. They can secrete catecholamines: noradrenaline (commonest), adrenaline, and dopamine. Catecholamines exert an effect on the alpha- and beta- receptors. Alpha1-receptor activation results in vasoconstriction causing systemic hypertension. Excess beta1-receptor activation leads to tachycardias and arrhythmias.
Prevalence
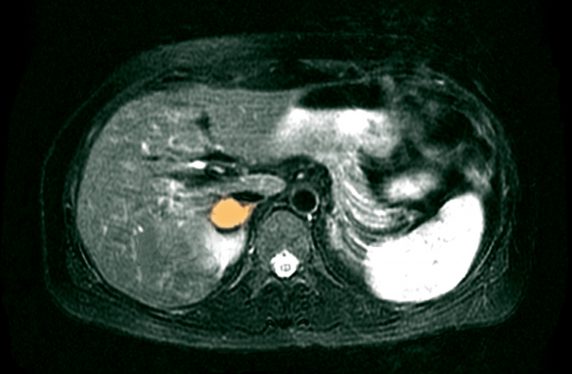
PCC is the more common tumour, with a reported incidence of 2-8 per million.1 Non-adrenal PGLs are rarer, with an incidence of 0.5 per million.2 The prevalence of phaeochromocytoma and paraganglioma (PPGL) in the hypertensive population is only 0.1-0.6%1 and only 4% of incidentally found adrenal lesions are PCCs.1,2 Up to 25% of PPGLs are discovered incidentally on imaging for unrelated disorders.3 The mean age at diagnosis is 43 years, with 10-20% occurring in children.3 PPGLs have been reported in patients as young as five, and younger age at diagnosis is more likely to be associated with an underlying genetic mutation.
Presentation
The classic symptom triad is palpitations, excess sweating and headaches but it has been reported that only 4% of patients actually present with the complete triad4. Patients with a secretory PPGL can present with a much wider range of other symptoms including anxiety, pallor, flushing, tremors, abdominal or chest pain, nausea, fatigue, weight loss, heat intolerance, visual disturbance, postural dizziness, change in bowel habit, neurological symptoms and an ‘impending sense of doom’ (the percentage of patients that present with these different symptoms is outlined in figure 1)2,5,6. The non-specific nature of these symptoms makes the diagnosis difficult in general practice, but the combination of hypertension with a cluster of any of these symptoms should raise one’s suspicion.
Additionally the presence of unusually severe or labile hypertension, hypertension with symptoms (figure 1), or the combination of new onset diabetes mellitus and hypertension, without the usual risk factors, especially in young non-obese patients,7 should also raise one’s index of suspicion. The diagnosis should always be ruled out in any patient with an incidentally identified adrenal lesion or a mass identified on imaging along the sympathetic and parasympathetic distribution. A lower threshold for testing should occur in any patient with a family history of PPGL disease or a carrier of a genetic mutation known to predispose to PPGL formation. Figure 1 shows the percentage of patients that report different symptoms.6
Clues in the history might be the episodic nature of the symptoms with complete resolution between severe episodes of a cluster of symptoms. The timings of the symptoms can be highly variable and not all patients report episodic symptoms. Some patients experience postural symptoms and a tachycardia can often be elicited on checking a postural blood pressure, due to catecholamine-induced volume contraction.
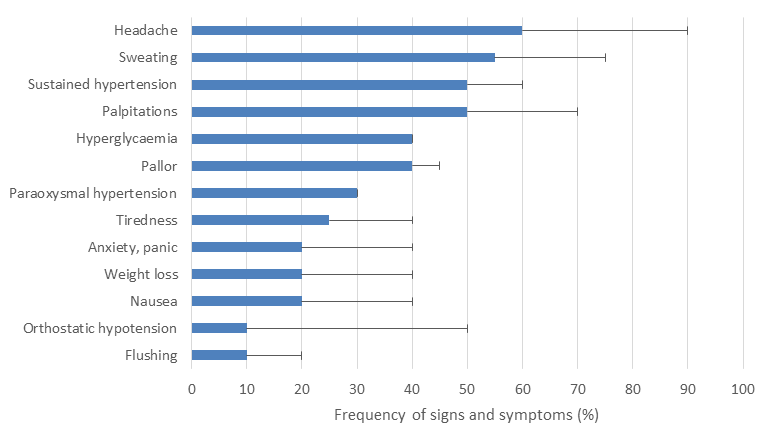
Figure 1: Common presenting symptoms. Bar chart showing the percentage of patients with phaeochromocytoma that reported different symptoms.2
Differential diagnosis
The historical difficulty in diagnosing PPGLs is that their presentation can mimic many other more common diseases, and the majority of clinicians may only see a few cases in the course of their careers. Patients with PPGLs can present to many different community, medical and surgical specialties. It is therefore imperative that all clinicians retain a high index of suspicion to ensure earlier detection of PPGLs, so that appropriate management can be commenced to avoid the associated morbidity and mortality. Table 1 shows a non-exhaustive list of potential differential diagnosis, where PPGL should be considered.
| Cardiovascular | Metabolic | Other |
|---|---|---|
|
Cardiomyopathy |
Thyrotoxicosis |
Alcohol withdrawal |
|
Unexplained arrhythmias |
An adrenal mass |
Use of illicit substances |
|
Hypertension in the young |
Carcinoid syndrome |
Anxiety/panic attacks |
|
Unexplained chest or back pain |
Hypoglycaemia |
Temporal lobe epilepsy |
|
Angina/IHD |
Difficult to control diabetes |
Menopause |
|
Heart failure |
Pre-eclampsia |
|
|
Renovascular disease |
Migraine |
Table 1: Non-exhaustive list of potential differential diagnosis where a diagnosis of PPGL should be considered.2
Investigation
Advances in biochemistry now make a secretory PPGL relatively easy to rule out, with a reliable and low-cost test in the form of a single normal plasma metanephrine/normetanephrine result or a single normal 24-hour urine collection for metanephrines (depending on local availability). Careful evaluation is needed of anyone with a result falling above the upper limit of the normal range. False negative results are unlikely, with the exception of a low result due to an incomplete 24 hour urine collection (it is essential that very clear instructions are therefore given to the patient, table 2). A plasma metanephrine assessment is more reliable for this reason as well as being more convenient for most patients. False positive results can however occur and any patient with raised levels of metanephrines should be discussed with an endocrinologist for consideration of further investigations.
Common medications that can cause mildly false positive results include regular use of paracetamol, alpha and beta-blockers, tricyclic antidepressants and monoamine oxidase inhibitors and sympathomimetics (e.g. nicotine, ephedrine, pseudoephedrine, cocaine).8,9
Abnormal results or a high index of suspicion should prompt an urgent referral to endocrinology so that alpha blockade can be safely commenced and localising tests undertaken.
| Biochemistry | How to take the samples |
|---|---|
|
Plasma metanephrines |
The patient needs to be fasted from midnight. Patient needs to be supine for 30 minutes before the blood test is taken (EDTA bottle). Sample needs to be centrifuged within two hours. This may require local arrangements to be set up. Patients should be advised to avoid taking any paracetamol or pseudoephedrine the day before and the morning of the blood test.
|
|
Urine metanephrines |
A complete 24 hour urine collection is required for accuracy. Patients should be advised Carefully on how to collect this. Urine samples can be collected into both acid and non-acid collection bottles. Urine should be collected into a separate container, and not into the collection bottle (if it contains acid), and then poured into the urine bottle. Patients should be advised to avoid taking any paracetamol or pseudoephedrine the day before and the day of the urine collection.
|
Table 2: Getting the correct samples.
Diagnosis
When the diagnosis is reached it is usually the case that symptoms have been present for several years and autopsy studies show higher prevalence rates suggesting many tumours still remain undetected in life.1 This highlights the importance of having a high index of suspicion and screening at-risk patients. The diagnosis is confirmed with successful localisation of the PPGL, with initial treatment being medical blockade in preparation for surgery if considered feasible. In 10-15% of cases, the PPGL can be malignant and in these cases, treatment options are more limited.
Follow up
Where possible, the aim will be curative surgery although stereotactic radiotherapy is increasingly recognised to be an option for head and neck PGL, carrying lower morbidity. It is now recognised that 40-50% of PPGL are due to underlying genetic mutations. Therefore all patients that present with PPGL are considered for genetic testing. If a causative mutation is identified, cascade genetic testing will be offered to family members. As a result of this cascade testing, there are increasing numbers of individuals that have been identified as carriers of genetic mutations that predispose them to developing PPGLs. There are now 16 recognised germline mutations that predispose to PPGL formation.10-12 The risk of developing a tumour and the type of disease will depend on the genetic abnormality. All of these patients should be reviewed in specialist centres to be offered appropriate surveillance screening.
Dr Nicola Tufton is a clinical research fellow in endocrinology, currently undertaking a PhD with the aim to improve the diagnosis and management of patients with phaeochromocytoma and paraganglioma.
Dr Scott Akker is a consultant endocrinologist at St. Bartholomew’s hospital, with a clinical practice specialising in adrenal, pituitary and thyroid disorders.
We acknowledge the input of Dr Lindsey Crockett, Dr Heather Noone and Dr Jane O’Donnell for reading a draft of this article and their helpful insights. Nicola Tufton is funded by The Medical College of Saint Bartholomew’s Hospital Trust.
References
- Corssmit EP, Romijn JA. Clinical management of paragangliomas. Eur J Endocrinol. 2014;171(6):R231-43.
- Lenders JW, Eisenhofer G, Mannelli M, et al. Phaeochromocytoma. Lancet. 2005;366(9486):665-75.
- Kirmani S, Young WF. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R). Seattle (WA)1993.
- Zhang R, Gupta D, Albert SG. Pheochromocytoma as a reversible cause of cardiomyopathy: Analysis and review of the literature. Int J Cardiol. 2017;249:319-23.
- Darr R, Lenders JW, Hofbauer LC, et al. Pheochromocytoma – update on disease management. Ther Adv Endocrinol Metab. 2012;3(1):11-26.
- Manger WM, Gifford RW. Pheochromocytoma. New York: Springer-Verlag; 1977. xxiv, 398 p. p.
- La Batide-Alanore A, Chatellier G, Plouin PF. Diabetes as a marker of pheochromocytoma in hypertensive patients. J Hypertens. 2003;21(9):1703-7.
- Neary NM, King KS, Pacak K. Drugs and pheochromocytoma–don’t be fooled by every elevated metanephrine. N Engl J Med. 2011;364(23):2268-70.
- Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-42.
- Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101-11.
- Martucci VL, Pacak K. Pheochromocytoma and paraganglioma: diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014;38(1):7-41.
- Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14(2):108-19.
Visit Pulse Reference for details on 140 symptoms, including easily searchable symptoms and categories, offering you a free platform to check symptoms and receive potential diagnoses during consultations.