Rheumatology clinic – temporal arteritis
Case
A 65-year-old lady presents to her GP complaining of bilateral temporal headache with shoulder girdle pain and stiffness for the past eight days. The symptoms have not responded to NSAIDs or common analgesics. She says that she has never experienced this type of headache previously. On direct questioning, she tells you that in the last two weeks she has noticed pain in her jaw and face, within a few minutes of starting to chew her food. This prompted her to visit her regular dentist who did not find any abnormalities. Her previous medical history includes arterial hypertension, well-controlled by treatment.
On examination she has a blood pressure of 120/70mmHg, she weighs 65kg (she has not lost any weight recently), is apyrexial, has a normal neurological examination and there is no evidence of cranial nerve palsy, nor any sign of shingles. She has no visual complaints. The temporal arteries are tender and swollen bilaterally. She cannot elevate her shoulders above her head, because of stiffness and pain.
Her GP suspects a diagnosis of giant cell arteritis (GCA), prescribes high dose glucocorticoid therapy (prednisolone 60mg daily) and urgently refers her to the fast-track GCA clinic at the rheumatology department. Here the clinical features are reviewed, she is examined and undergoes colour Doppler sonography (CDS) of the temporal and axillary arteries showing features suggestive of GCA (figure 1). The diagnosis of GCA is confirmed and treatment with glucocorticoids is continued.
The problem
GCA is an autoimmune disorder that predominantly affects older people (>50 years), causing chronic inflammation of medium to large sized arteries, usually affecting the aorta or its major branches. The condition, formerly known as Horton’s disease, or temporal arteritis, was officially redefined as GCA in 2012 during a consensus conference to review the nomenclature of all forms of primary vasculitis.1 The trigger for this immune dysregulation is not well understood. The arterial inflammation results in narrowing and blockage and therefore has significant morbidity, particularly when it manifests itself in the form of stroke, and optic nerve ischaemia leading to permanent visual loss.2 The disease spectrum includes overlapping phenotypes including classic cranial arteritis or extra-cranial GCA, defined as large-vessel GCA, which can occur in up to 83% of cases.3 The involvement of extra-cranial branches can be complicated by the development of aortic aneurysms and dissection.4
GCA is the most common form of primary vasculitis, predominantly affecting women, with a peak incidence at 70-80 years of age. The incidence is highest in white Caucasians in Northern Europe (20 cases per 100,000 persons older than 50 years).5 A recent study reported a prevalence of 0.41% for GP-diagnosed cases of GCA (patients who were treated with glucocorticoids and in whom the diagnosis was not later refuted) in a UK primary care population over a period of 30 years.6
The presentation of patients with recent onset headache with features suggestive of GCA such as jaw claudication, polymyalgia rheumatica (PMR), characterised by inflammatory pain and stiffness of the neck, shoulder and hip girdles and raised inflammatory markers, combined with systemic symptoms such as weight loss or fever should strongly suggest a diagnosis of GCA.
GCA is considered a medical emergency, because of the risk of sudden irreversible sight loss.7 When suspected, treatment should be initiated immediately and patients should be promptly referred to on-call disciplines such as rheumatology, or when not available, ophthalmology or a medical emergency department.
Features
GCA typically presents with headache that occurs in two thirds of patients. The pain is more often temporal or occipital, but may be less well defined8, and can be associated with scalp tenderness. The temporal arteries may be thickened, nodular and tender with reduced or absent pulses. Symptoms of jaw claudication occur in about half of all patients at presentation and occasionally claudication affects the tongue.8,9 Visual loss occurs in 10-15% of patients and is usually caused by ischaemic optic neuropathy.10 Blindness in GCA is generally irreversible and may be preceded by amaurosis fugax, diplopia and jaw claudication.3,9,10 If the patient is left untreated, the other eye is likely to become affected within a few days or weeks.11,12 Less frequently, stroke, limb claudication and tongue and scalp necrosis may occur, with less common features including cough, sore throat, and hoarseness.8,11,12 Up to 50% of patients have constitutional symptoms (fever, malaise, weight loss), particularly in large vessel GCA. PMR is reported in 40-60% of patients with GCA at onset, and 16-21% of patients with PMR will develop GCA. PMR is also the most frequent symptom in case of GCA relapse (50% of cases).
Diagnosis
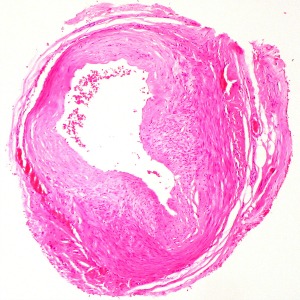
The diagnosis of GCA is mainly clinical. Until recently, the gold standard for diagnosis has been temporal artery biopsy (TAB), but this can be negative in up to 60% of patients. Giant cells are typically present in around 70% of biopsies, but are not necessary for diagnosis as long as there is evidence of significant inflammation in the adventitia, media or intima. Colour Doppler ultrasound of temporal arteries and large vessels is an emerging diagnostic tool for GCA.13 Ultrasound examination of large vessels can provide information about the presence of vessel wall oedema, known as a ‘halo’ (figure 1), throughout the length of the vessel, potentially overcoming the problem of skip lesions often affecting the results of histological examination.15 18 fluorodeoxyglucose-positron positron emission tomography combined with computed tomography (FDG PET-CT) or magnetic resonance angiography can be useful to detect extra-cranial large-vessel involvement. There is no specific laboratory test to confirm GCA. Acute phase reactants are usually elevated, nevertheless, up to 20% of patients with GCA have normal ESR before treatment (especially those patients who are at risk of sight loss); CRP is more sensitive but a normal level does not exclude the diagnosis. The two tests together reach a sensitivity of 99%.16 Further routine investigations for suspected GCA are presented in the table below.
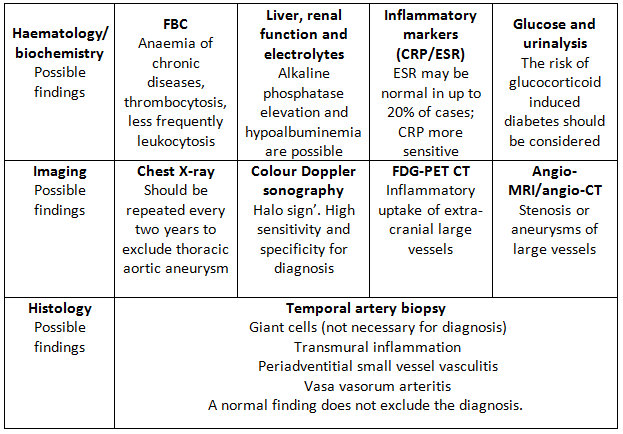
Table 1
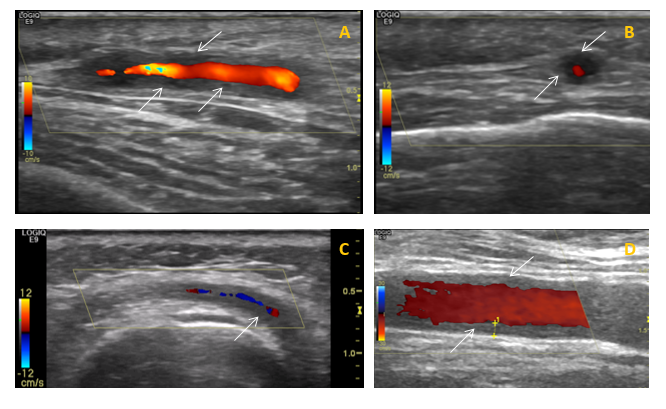
Figure 1
Management
Glucocorticoids represent the cornerstone of GCA treatment and should be started as soon as GCA is clinically suspected to prevent permanent sequelae such as visual loss.17 The British Society of Rheumatology (BSR) guidelines18 suggest to prescribe prednisolone 40-60mg daily in uncomplicated GCA (no jaw claudication or visual disturbance). In complicated GCA (evolving visual loss or amaurosis fugax) treatment with 500mg to 1g of intravenous methylprednisolone for up to three days should be considered in addition to oral glucocorticoids. In cases of established visual loss 60mg of prednisolone daily is recommended to protect the contralateral eye. The initial high dose of oral glucocorticoids should be continued for at least 3-4 weeks (invariably until symptoms and laboratory abnormalities resolve) a very slow tapering regimen can then be started. In cases of recurrent relapse or failure to reduce the glucocorticoid dose, an adjuvant immunosuppressive therapy (e.g. methotrexate) should be considered. Low-dose aspirin should be considered if not contraindicated, although its role in preventing ischaemic complications is still uncertain. Patients should receive bone protection. PPIs can be considered. Recent promising evidence suggests that biological therapies (particularly IL-6 inhibitors) are effective in GCA.17
Professor Raashid Luqmani is a consultant rheumatologist at Oxford University Hospitals NHS Trust
Dr Sara Monti is a specialist registrar at the University of Pavia, Italy
Dr Cristina Ponte is a clinical research fellow at the University of Oxford and University of Lisbon
Visit Pulse Reference for details on 140 symptoms, including easily searchable symptoms and categories, offering you a free platform to check symptoms and receive potential diagnoses during consultations.






