Neurology clinic – Charcot Marie Tooth disease
A 21-year-old man presents to his GP with a six-month history of tingling and numbness in his feet. A detailed history reveals that he has a longstanding history of a tendency to sprain his ankles, has always been clumsy and has never been a fast runner. He has always had high arches, and says he turns his feet in when walking. In his family, his father had similarly shaped feet.
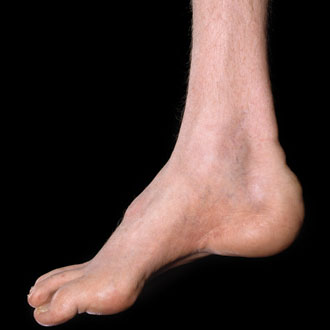
On examination, he has pes cavus and hammer toes. He is able to stand on his toes but not on his heels, and has bilateral Achilles tendon tightness. There is wasting of his distal upper and lower limbs and he has mild distal weakness in the lower limbs with ankle dorsiflexion weakness bilaterally. He is areflexic. Sensory examination shows normal sensation in the upper limbs but reduction of pin-prick reaction in the feet up to the ankles. Vibration sense is reduced in both ankles.
The GP suspects a peripheral neuropathy and, because of the longstanding symptoms and the possible relevant family history, refers him to a consultant neurologist.
The problem
Charcot Marie Tooth disease (CMT), also known as hereditary motor and sensory neuropathy, is a clinically and genetically heterogeneous group of sensorimotor peripheral neuropathies. CMT has now been recognised as the most common inherited neuromuscular disorder, affecting at least one in 2,500 individuals.1 Patients with CMT are traditionally classified into three main groups based on upper limb (median or ulnar) motor nerve conduction velocity (MNCV), and these guide genetic testing:
• MNCV <38m/s is considered demyelinating and classifies a patient as having CMT1.
• Patients with CMT2 have MNCV >38m/s and often have small compound motor action potentials (CMAP) indicating axonal loss.
• A third group has MNCV 25-45m/s and may be classified as intermediate CMT.
Patients may have autosomal dominant, autosomal recessive or X-linked inheritance, but de novo dominant cases are not rare and therefore some patients do not have a family history.
Features
CMT is characterised by predominant distal muscle wasting and weakness affecting the legs more than the arms. Sensory loss in a glove and stocking distribution and reduced or absent tendon reflexes are also common. Foot deformities (pes cavus, hammer toes) and scoliosis are frequently observed. The onset occurs usually in the first two decades of life. The disease course is generally slowly progressive.
Diagnosis
A detailed history is important to differentiate acquired versus inherited causes of neuropathy. Although patients may complain of symptoms for only a relatively short time, detailed questions regarding their childhood and development may reveal a much longer history. Parents may recall that the patient walked later than their siblings, always had high arches and difficulty fitting shoes. Patients commonly recall being poor at sports, coming last in races and have a tendency to trip and sprain their ankles. Women may have had difficulty wearing high heels. A detailed family history is essential to determine pattern of inheritance.
A careful examination should be performed to identify muscle wasting and weakness, sensory loss and foot deformities.
Neurophysiology is essential in the diagnosis and classification of CMT. Nerve-conduction studies show abnormalities of both motor and sensory nerves, and electromyography typically shows evidence of denervation in a length-dependent pattern.
Currently, the majority of patients with CMT1 obtain a genetic diagnosis, but only around 35% of patients with CMT2 are diagnosed this way.
When to consider inherited neuropathy
• Longstanding history
• Slowly progressive symptoms
• Foot deformities
• Family history
• Positive sensory symptoms less common
Management
Currently there is no pharmacological treatment for any form of CMT, but disease-specific treatment is emerging in clinical trials. Referral to a specialist neurology clinic is essential for diagnosis and management of the condition. Management is currently focused on physiotherapy, use of orthoses, exercise, orthopaedic interventions for foot deformities and scoliosis, pain and fatigue management. It also includes genetic counselling for diagnostic, predictive, prenatal and for pre-implantation diagnosis.2
Prognosis
Patients have normal life expectancy. CMT is a chronic, slowly progressive disease whose severity can vary greatly among individuals even within the same family.
Dr Matilde Laurá is a consultant neurologist at the MRC Centre for Neuromuscular Diseases
Professor Michael Hanna is a consultant neurologist and director of the MRC Centre for Neuromuscular Diseases at the UCL Institute of Neurology
Visit Pulse Reference for details on 140 symptoms, including easily searchable symptoms and categories, offering you a free platform to check symptoms and receive potential diagnoses during consultations.