Respiratory clinic – non-productive cough and joint pain
A 29-year-old, non-smoking man presents to his GP with a two-week history of acute onset joint pains particularly affecting the ankles, wrists and knees, a tender blotchy rash over his shins, intermittent fever, and a mild non-productive cough.
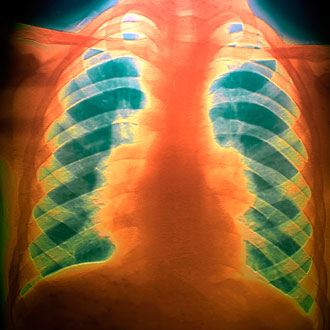
On examination, he is febrile with a temperature of 37.8 degrees Celsius, has moderate swelling and tenderness of the large joints, and cutaneous changes over the shins consistent with erythema nodosum. Examination of the respiratory system is unremarkable. A subsequent chest radiograph discloses bilateral hilar adenopathy and bilateral upper-zone nodular parenchymal infiltrates. The GP suspects sarcoidosis, treats with NSAIDs, and refers to a respiratory physician for assessment.
The Problem
Sarcoidosis is an enigmatic disease, the cardinal feature of which is non-caseating granulomatous inflammation, typically affecting the lungs and lymph nodes but potentially involving any organ in the body. The condition most commonly occurs in those in their fourth and fifth decades and shows an equal gender split.1 The incidence rate of sarcoidosis in the UK is five per 100,000 per year.2
Loefgren’s syndrome is an acute onset variant of sarcoidosis characterised by the triad of erythema nodosum, arthralgia and hilar adenopathy on chest X-ray. The Loefgren’s variant of sarcoid occurs most commonly in individuals with a Scandinavian heritage and is associated with genetic polymorphisms in specific human leukocyte antigen (HLA) genes. The condition, in contrast to some other forms of sarcoidosis, has an excellent prognosis and typically resolves fully within two years of onset.
Features
Sarcoidosis may also manifest with chronic, progressive disease, which has the potential to involve any organ. Varying patterns of organ involvement result in the condition causing diverse symptoms in different individuals. To add to this complexity, sarcoidosis may also be asymptomatic and therefore be diagnosed incidentally.
Over 95% of those with sarcoidosis have pulmonary involvement. Consequently, the most common presenting symptoms are cough or breathlessness. Other common symptoms relate to ocular (usually uveitis) and cutaneous disease – both of which occur in approximately 15% of patients – and to hypercalcaemia or hypercalciuria, which occur in about 10%. Heerfordt’s syndrome (also known as uveoparotid fever) is another acute onset form of the disease and presents with uveitis, parotitis (often causing compression and thus paralysis of the facial nerve) and fever.
Diagnosis
In general, the diagnosis of sarcoidosis is one of exclusion, which relies on the combination of appropriate clinical presentation together with the demonstration of non-caseating granulomata on tissue biopsy. It is important that other causes of granulomatous inflammation (the commonest of which is tuberculosis infection) are excluded. Chest X-ray in sarcoid commonly demonstrates hilar or mediastinal adenopathy, often with associated parenchymal infiltrates. Serum angiotensin converting enzyme, when elevated (which is in about a third of cases), is a useful measure of disease activity and, similarly, a raised serum calcium often provides an important pointer to diagnosis. In the acute onset variants of the disease, for example Loefgren’s and Heerfordt’s syndromes, the constellation of symptoms combined with typical imaging findings (hilar adenopathy on chest radiograph) are sufficient to confirm a diagnosis of sarcoid. However, a biopsy of the most accessible affected organ is often necessary to confirm the presence of non-caseating granulomatous inflammation and to exclude alternate diagnoses. Cardiac involvement in sarcoidosis can be potentially life-threatening. In cases where sarcoidosis is suspected or confirmed, potential cardiac symptoms – such as palpitations or syncope – merit urgent assessment in a multidisciplinary cardiac-sarcoid unit. Profound fatigue is a common, and often underappreciated, symptom in all forms of sarcoidosis.
Management
In the majority of cases, sarcoidosis is self-limiting with an excellent prognosis, and so treatment is both only required to manage intrusive symptoms, and often achievable with NSAIDs. But a significant minority of patients have a progressive disease that leads to the development of end-organ fibrosis. In some cases, granulomatous inflammation can acutely threaten organ function (for example, in those with ocular or cardiac involvement). In such cases, treatment is vital to prevent life-shortening or irreversible complications.
Corticosteroids form the basis of most therapeutic regimens for sarcoidosis while azathioprine, methotrexate or infliximab are used in refractory disease, albeit with a very limited evidence base.3,4 The choice and duration of the treatment is driven by expert consensus, and it remains to be demonstrated whether therapy in sarcoidosis modifies disease progression.
Conclusion
The multi-faceted nature of sarcoidosis makes it a difficult disease to diagnose and treat. There is an urgent need for clinical trials to address questions surrounding which patients to treat, when, and for how long. For the majority of patients, sarcoidosis is a self-limiting disease that does not affect life expectancy. However, for those unlucky enough to develop progressive disease with end-organ fibrosis, sarcoidosis can be both debilitating and life-shortening. Such patients should be under the regular care of dedicated sarcoidosis services, with access to multi-specialty clinical teams.
Dr Toby M Maher is consultant respiratory physician at the Royal Brompton Hospital, London.
References
1. Baughman RP, Teirstein AS, Judson MA et al. Case Control Etiologic Study of Sarcoidosis (ACCESS) research group. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med, 2001;164:1885-89
2. Gribbin J, Hubbard RB, Le Jeune et al. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax, 2006;61:980-85
3. Paramothayan NS, Lasserson TJ, Jones PW. Corticosteroids for pulmonary sarcoidosis. Cochrane Database Syst Rev, 2005;18:CD001114.
4. Paramothayan S, Lasserson TJ, Walters EH. Immunosuppressive and cytotoxic therapy for pulmonary sarcoidosis. Cochrane Database Syst Rev, 2006;3:CD003536.
Visit Pulse Reference for details on 140 symptoms, including easily searchable symptoms and categories, offering you a free platform to check symptoms and receive potential diagnoses during consultations.